《表1 BDCPDT的光物理性质》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
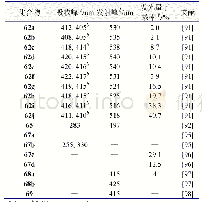
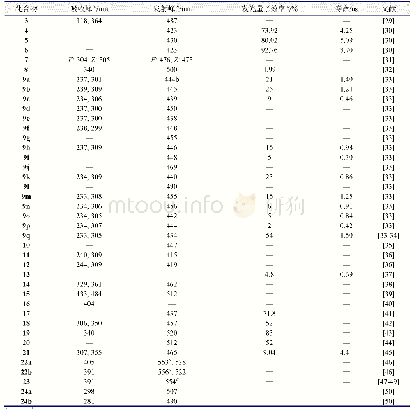
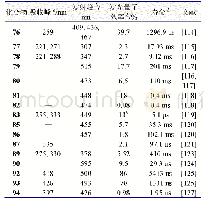
结构决定性质,为了更清楚的探究最终合成产物的结构,进一步了解化合物结构特征和光物理性质之间的相关性,用Gaussian 09软件对目标产物进行密度泛函理论(DFT)计算。在B3LYP方法下,以6-31G(d,p)基组获得的BDCPDT的优化结构、最高占据轨道(HOMO)和最低空轨道(LUMO)的电子云分布如图5所示。在图5中可以看出,BDCPDT分子是以四苯基乙烯(TPE)为核心结构,咔唑作为外围基团,且整体呈螺旋状排列,即四苯乙烯基团和咔唑基团处于不同平面,这种构型有效地消除了分子间紧密的π-π堆积,从而产生聚集诱导发光(AIE)性能;BDCPDT的HOMO轨道主要分布在外围咔唑和与其相连的苯环上,LUMO轨道主要分布在四苯乙烯基团上,HOMO和LUMO的交叠部分位于四苯基乙烯和咔唑相连的苯环上,这有利于分子内电荷转移。计算可得BDCPDT的HOMO能级为-5.43eV,LUMO能级为-1.67eV,HOMO和LUMO的能隙为3.76eV。相关数据总结在表1中。
| 图表编号 | XD0057581600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.05.15 |
| 作者 | 张文娟、王洪波、刘少洲、赵耀东、白生弟、施和平 |
| 绘制单位 | 山西大学化学化工学院、山西大学化学化工学院、山西大学化学化工学院、山西大学化学化工学院、山西大学应用化学研究所、山西大学化学化工学院 |
| 更多格式 | 高清、无水印(增值服务) |

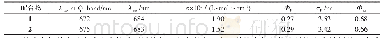

{kind=link}