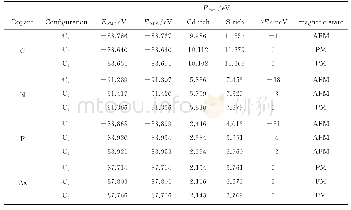
《表4 B3LYP/def2-TZVP水平下不同配合物计算HS、BS态能量 (E) 及磁耦合常数J值》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《羧酸桥对反铁磁性EO叠氮铜双核配合物磁性影响的理论研究》
a:L1=N-(3-aminopropyl)salicylaldimine;b:L2=7-amino-4-methyl-5-azahept-3-en-2-one;c:L3=8-amino-4-methyl-5-azaoct-3-en-2-one;d:2,6-bis[{[(2-hydroxybenzyl)(N,N-(dimethylamino)ethyl]amino}methyl]-4-methylphenol.
为验证计算条件的可靠性,选择9个不同磁学性质的叠氮铜双核配合物进行计算,结果如表4所示.从表4可以看出,无论是反铁磁性耦合的体系,还是铁磁性耦合的体系,在B3LYP/def2-TZVP水平下采用密度泛函理论结合对称性破损态(DFT-BS)方法计算叠氮铜双核配合物的磁学性质得到的结果与实验测量的磁学性质一致.所以选择该条件研究叠氮铜双核配合物磁学性质是适合的,也可用于预测新合成的叠氮铜双核配合物的磁学性质.
| 图表编号 | XD0018989200 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2018.08.01 |
| 作者 | 罗树常、李宏媛、郑鹏飞、孙小媛、刘红、刘翔宇、李佐 |
| 绘制单位 | 贵州工程应用技术学院化学工程学院、贵州工程应用技术学院化学工程学院、贵州工程应用技术学院化学工程学院、贵州工程应用技术学院化学工程学院、贵州工程应用技术学院化学工程学院、宁夏大学化学化工学院、贵州工程应用技术学院理学院 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}