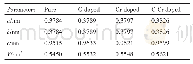
《表1 优化的面心立方晶胞MxCu1-x和体心立方晶胞M1Cu1的晶格参数》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《单原子合金催化剂热稳定性的反应力场分子动力学研究》
采用基于密度泛函理论(DFT)的从头量子力学的程序包VASP(Vienna Ab-initio Simulation Package)[21]进行计算.基组采用平面波赝势方法[22],利用PBE关联梯度修正泛函对模型进行充分优化[23],采用广义梯度近似(GGA)描述体系的交换关联能.设置平面波截断能400eV,k点[24]为3×3×1.计算含有Fe和Ni的体系时,加入了自旋极化校正.晶胞厚度为4层,扩胞3×3.负载型单原子催化剂模型(M1/Cu(111))为添加一个额外的原子在fcc或hcp位点,而掺杂型原子催化剂模型(M1@Cu(111))为取代表层的一个Cu原子.在优化过程中,上两层弛豫,下两层原子保持固定.为优化反应力场参数,优化了MxCu1-x(x=0.25,0.5,0.75)合金的晶格参数(见表1).此外,本文研究中采用CI-NEB[25]方法来搜索过渡态.
| 图表编号 | XD00140459600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.04.10 |
| 作者 | 王贵昌、杨文琦、汪杰、乔园园 |
| 绘制单位 | 南开大学化学学院、先进能源材料化学教育部重点实验室、化学科学与工程协同创新中心(天津)、南开大学化学学院、先进能源材料化学教育部重点实验室、化学科学与工程协同创新中心(天津)、南开大学化学学院、先进能源材料化学教育部重点实验室、化学科学与工程协同创新中心(天津)、南开大学化学学院、先进能源材料化学教育部重点实验室、化学科学与工程协同创新中心(天津) |
| 更多格式 | 高清、无水印(增值服务) |




![表3 面心立方FCC金属剪切织构及其对应的及Δr值[16]](http://bookimg.mtoou.info/tubiao/gif/JSRC202012015_03600.gif)
{kind=link}