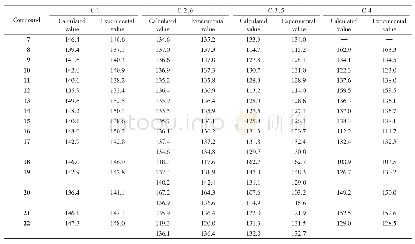
《表7 化合物1~4的碳原子化学位移量化计算》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
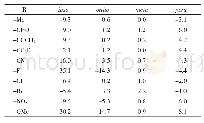
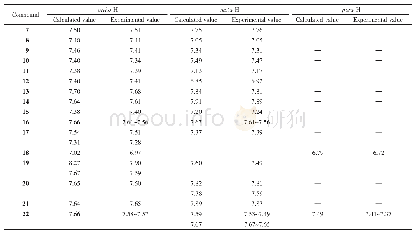
2.5.3化合物1~4化学位移的量化计算本文使用6-311+G(2d,p)基组,运用B3LYP密度泛函对相关分子结构进行了优化,并使用PCM溶剂化模型考虑了CDCl3的影响,随后的1H NMR和13C NMR计算使用了与优化水平相同的计算水准和溶剂化模型。化学位移计算时使用了GIAO和CSGT两种方法,以TMS的氢和碳的化学位移为基准,全部计算使用Gaussian 09程序完成。结果(表6和7)表明,数据均在计算误差范围之内,并且两种方法(GIAO和CSGT)计算的13C NMR结果相近。
| 图表编号 | XD00119071400 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.01.18 |
| 作者 | 刘季红、靳焜、田镇豪、罗根 |
| 绘制单位 | 大连理工大学化工学院、大连理工大学精细化工国家重点实验室、大连理工大学精细化工国家重点实验室、大连理工大学化工学院 |
| 更多格式 | 高清、无水印(增值服务) |



{kind=link}