《表1 MoP(001)表面上哌啶的吸附位、吸附能及相关结构参数》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
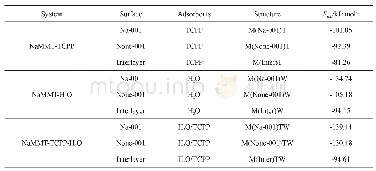
首先优化气相哌啶的分子结构,如图2所示,计算得到的键长为0.109 9~0.111 2 nm(C—H键)和0.102 0 nm(N—H键)。考虑到哌啶环的饱和特性,可以推测哌啶只能通过N的孤对电子与表面发生作用从而形成稳定吸附。计算结果证实哌啶只能通过N—Mo键吸附在Mo原子顶位。如图3所示,哌啶在MoP(001)表面上存在4种稳定的吸附构型,依次标记为hcp、fcc、top-1、top-2,分别表示哌啶环位于hcp、fcc和top位的上方。表1给出了这些构型对应的吸附能及重要的结构参数。吸附能分别为0.67 eV(hcp)、0.66 eV(fcc)、0.61 e V(top-1)和0.60 eV(top-2)。哌啶在MoP上较弱的吸附与其较长的N—Mo键(均大于0.230 nm)相吻合。此外,哌啶环中的相关键长并未因吸附而发生没有明显的改变,可认为C—N键并未受到活化作用。
| 图表编号 | XD00111797100 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.09.01 |
| 作者 | 朱后禹、匙玉华、赵联明、徐静、郭文跃 |
| 绘制单位 | 中国石油大学(华东)材料科学与工程学院、中国石油大学(华东)材料科学与工程学院、中国石油大学(华东)材料科学与工程学院、中国石油大学(华东)材料科学与工程学院、中国石油大学(华东)材料科学与工程学院 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}