《表1 Li7MB8H32 (M=Li、Fe、Co和Ni) 体系的晶胞参数及能量计算值》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《Fe、Co和Ni掺杂LiBH_4放氢性能的第一性原理研究》
常温下,Li BH4具有正交结构,空间群为Pnma,晶胞参数为a=7.179、b=4.437、c=6.803,原子坐标为Li(4c)(0.157、0.25、0.102) 、B(4c)(0.304、0.25、0.431) 、H1(4c)(0.9、0.25、0.956) 、H2(4c)(0.404、0.25、0.28) 和H3(8d)(0.172、0.05、0.428) [19]。对上述Li BH4晶胞进行几何结构优化,所得结果(a=7.255、b=4.398、c=6.727)(表1) 与实验测试(a=7.179、b=4.437、c=6.803[19])及理论计算值(a=7.32、b=4.37、c=6.59[20])非常接近。将优化的Li BH4晶胞沿b轴延伸建立1×2×1超胞模型(a=7.255、b=8.795、c=6.727),如图1所示,该超胞模型包含8个Li原子、8个B原子和32个H原子,标记为Li8B8H32(M0-Li)。由图1可见,Li BH4晶胞内1个B原子周围围绕4个氢原子(HA、HB、HC和HD)组成[BH4]单元。通过Fe、Co、Ni分别置换图1中Li原子(4c)(0.338、0.375、0.613)(标记为S1),构建Fe、Co、Ni掺杂体系Li7Fe B8H32(M0-Fe)、Li7CoB8H32(M0-Co)、Li7Ni B8H32(M0-Ni)。对掺杂体系晶胞结构进行几何优化,所得结果列于表1。很显然,与本体M0-Li体系晶胞参数相比,Fe、Co、Ni添加使体系晶胞略沿b轴伸展、沿c轴收缩,这源于掺杂元素Fe(1.17)、Co(1.16)和Ni(1.15)的原子半径与被掺杂元素Li的原子半径(1.23)存在差异。为便于分析,本工作中以Li7MB8H32(M=Li、Fe、Co和Ni)代表所有研究体系Li8B8H32(M0-Li)、Li7Fe B8H32(M0-Fe)、Li7CoB8H32(M0-Co)和Li7Ni B8H32(M0-Ni)。
| 图表编号 | XD0029108800 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.01.25 |
| 作者 | 莫晓华、蒋卫卿 |
| 绘制单位 | 广西民族大学理学院电离层观测与模拟重点实验室、广西大学物理科学与工程技术学院广西电化学能源材料重点实验室培育基地 |
| 更多格式 | 高清、无水印(增值服务) |
![表1 La Mg X4(X=Co,Ni,Cu)的晶体学参数、晶格常数a、晶胞体积V0、结合能Ec、形成焓ΔH,并与其它计算值比较[16,17,27-29]](http://bookimg.mtoou.info/tubiao/gif/YZYF202102023_01700.gif)

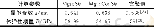



{kind=link}