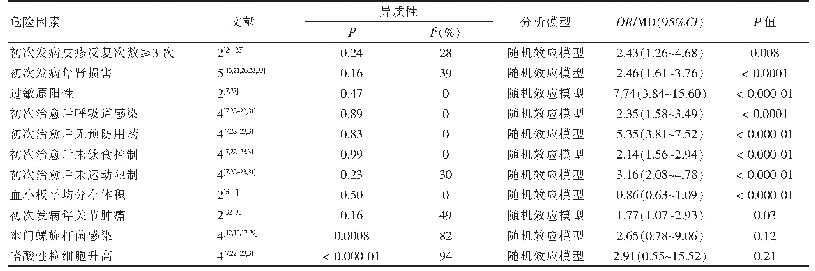
《表2 HSP的发病机制》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
注:参考文献[6,21-23]
PLP1(SPG2型)编码的PLP1蛋白,是中枢神经系统中最为丰富的内源性髓鞘蛋白,具有促进髓鞘成熟和维持膜结构稳定的功能。Luders等[18]在小鼠模型中证明,PLP1突变致少突胶质细胞功能障碍,为SPG2轴索退行性变的主要原因。此外,AP-4复合物由4种亚单位(β4、ε、μ4和σ4)组成,4种亚单位分别由不同基因编码,可导致不同类型的HSP:β4(AP4 B1,SPG47)、ε(AP4 E1,SPG5 1)、μ4(AP4 M1,SPG5 0)和σ4(AP4 S1,SPG5 2)。AP-4可促进自噬相关蛋白ATG9 A从反式高尔基体网络到周围细胞质的运输,有利于自噬蛋白LC3 B的脂化和自噬体的成熟,提示AP-4参与自噬功能的调节,其缺乏导致自噬功能改变,HSP疾病的发生[19]。同时,Vantaggiato等[20]发现在ZFYVE2 6(SPG1 5型)突变患者的淋巴母细胞和成纤维细胞中,自噬体成熟障碍及未成熟自噬体增加,推断其可能与SPG15的发病有关。除了以上描述的发病机制外,其它HSP相关发病机制。见表2。
| 图表编号 | XD0022076900 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2018.10.28 |
| 作者 | 曹丽荣、赵澎、蔡春泉 |
| 绘制单位 | 天津医科大学研究生院、天津市儿童医院康复科、天津市儿童医院神经外科 |
| 更多格式 | 高清、无水印(增值服务) |



![表1 儿童组和成人组合并肾损害HSP患者的发病特点和可能诱因[n(%)]](http://bookimg.mtoou.info/tubiao/gif/YYLC202003047_01200.gif)
{kind=link}