《表1 空白组和模型组IL-1β、TNF-α、NF-κB含量比较(±s)》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
现代研究指出,尽管可控性炎症具有一定的抗肿瘤效应,尤其是免疫系统中的T细胞介导的免疫应答反应会及时地清除癌细胞,使机体免于肿瘤的侵害[5-6],但非可控性炎症却能诱导肿瘤的发生和肿瘤的发展。非可控性炎症的主要病理变化是局部组织的变质和渗出、增生,在非可控性炎症的发展过程中,除了会激活大量炎性细胞,如肥大细胞、树突状细胞、巨噬细胞等,还会释放大量炎性介质,诸如细胞因子、生长因子、趋化因子等,以上炎性介质能够促进炎性微环境的形成,炎性介质可以趋化大量的炎性细胞,加剧非可控性炎症,导致恶性循环。非可控性炎症存在时,炎性微环境中的大量细胞因子、生长因子等炎性因子长期存在,导致DNA氧化损伤、造成基因组的不稳定,激活原癌基因、抑制抑癌基因的表达,促使肿瘤的发生[7-9]。促炎细胞因子是炎症反应的代表[10],其中IL-1β是重要的促炎细胞因子,该因子可以直接激活NF-κB,造成炎症级联反应,促使IL-6、IL-8、TNF-α的表达,不仅如此IL-1β还能增加IL-1βmRNA的产生,增加相关蛋白的转录,研究证明该过程是通过IL-1β核转录的作用进行的[11]。非可控性炎症持续存在导致IL-1β持续高水平,导致上皮细胞DNA突变酶表达异常,促进致癌通路的激活。PLGC正常上皮细胞恶变的必须条件是核苷酸改变和染色体异位等基因异常,IL-1β通过ATD的活性诱导DNA序列发生变化,从而实现上述基因异常,导致胃黏膜萎缩,加剧PLGC的发生和恶变[12-13]。除此以外,Hp感染后IL-1β等促炎细胞因子在胃黏膜中高度表达,IL-1β基因多态性影响Hp感染后IL-1β的分泌量,能够大幅度抑制胃酸的分泌,而Hp感染后胃黏膜IL-1βmRNA的表达增加,IL-1β分泌增多,造成恶性循环,增加胃黏膜萎缩和发生胃癌的风险[14-16]。TNF-α也是重要的炎症因子之一,其由巨噬细胞和活化T细胞分泌,该因子有多种生物学活性,能够在炎症和免疫应答的机制中起到重要作用[17]。TNF-α致癌的机制尚不明确,大多认为高水平的TNF-α能够通过NF-κB激活诱导的ATD的产生,引起肿瘤相关基因的突变,也能够通过多种信号通路导致上皮-间质转化、单核细胞-内皮细胞表型转变,促进免疫逃逸等起到促癌的作用[18-21]。在非可控性炎症起到重要作用的NF-κB是癌变过程的主要参与因子之一,其所参与的炎症信号下游级联式反应可能使胃黏膜出现癌变,有研究指出胃癌组织中的NF-κB通路呈过表达状态,其调控的Bcl-2、IL-8等都参与胃癌的发生发展[22]。其中p65是NF-κB的重要组件之一,NF-κB-p65磷酸化之后进入细胞核,促进靶基因转录,研究证明细胞核与细胞质中的NF-κB-p65的表达与胃癌的临床发展相关[23]。我们之前的实验[24]观察到PLGC的大鼠体内炎症因子的含量明显高于正常大鼠。我们运用改良MNNG+复合法造PLGC的大鼠模型,实验造模成功后分别予中药和西药观察炎症因子的变化,模型组与空白组的大鼠比较发现IL-1β、TNF-α、NF-κB水平较正常组大鼠明显升高(P<0.05),如表1。这些炎症因子从多种通路导致非可控性炎症持续并且加重,影响血管新生和细胞凋亡,促进癌细胞的增殖,提高癌细胞的侵袭能力[25-27]。结合非可控性炎症在肿瘤发生发展中的作用,考虑非可控性炎症中以上几个炎症因子参加了PLGC的发生过程,并且对PLGC的发展有重要作用。经过治疗后高剂量组和西药组的各项指标均有明显下降,提示药物治疗能够有效地改善非可控性炎症的发展。基于非可控性炎症在PLGC中的机制,目前已有部分药物通过实验研究被证实有逆转胃黏膜萎缩、肠化和不典型增生的效果[28]。
| 图表编号 | XD00110034900 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.09.28 |
| 作者 | 马志豪、祁向争 |
| 绘制单位 | 天津中医药大学、天津中医药大学第二附属医院 |
| 更多格式 | 高清、无水印(增值服务) |


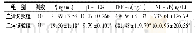



{kind=link}