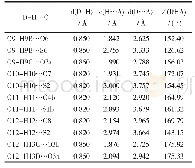
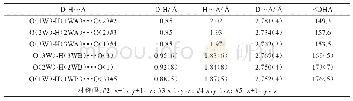
《表4 全构型优化理论计算及实验晶体解析得到的配合物的部分键长和键角》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
全构型优化计算中,没有任何对称性限制,通过对频率的计算,得到的谐振频率都是正值,说明优化得到的构型是能量极小值。表4为配合物全优化后基态构型的主要几何参数和实验数据对比。从表中可以看出,理论计算结果除Co—NCN的键长(2.186)稍大于实验结果(2.053)0.13外,其他键长与键角基本相符。计算值和实验值的差异主要是因为,量子化学计算模型是孤立状态下的气态结构,而实验值是晶体,存在分子间作用力。因此,在误差范围内,可以认为采用密度泛函理论的BP86/6-31G*+SDD方法能够得到配合物的准确分子结构。
| 图表编号 | XD0066033200 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.07.15 |
| 作者 | 刘婷、姚瑶、金栋女、金杰、魏荣敏 |
| 绘制单位 | 德州学院化学化工学院山东省功能材料与配位化学高校重点实验室、德州学院化学化工学院山东省功能材料与配位化学高校重点实验室、德州学院化学化工学院山东省功能材料与配位化学高校重点实验室、德州学院化学化工学院山东省功能材料与配位化学高校重点实验室、德州学院化学化工学院山东省功能材料与配位化学高校重点实验室 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}