《表1 HL1~HL3的理论计算数据》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
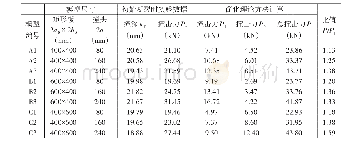
图2和表1为HL1~HL3的分子轨道能级和理论计算数据。从图2和表1中可以看出,HL1的理论计算最大吸收峰位于354 nm,主要来自HOMO到LUMO+2的跃迁,可归属于ICT跃迁。在短波长区,实验测定的最佳吸收峰位于289 nm,与理论值292 nm基本一致,是HOMO-1到LUMO+2的跃迁吸收,可归属为三苯胺的π-π*跃迁。HL2在363 nm处的吸收峰为HOMO到LUMO+2的跃迁,可归属于ICT跃迁。287 nm处的吸收峰对应于HOMO到LUMO+5,可归属为三苯胺的π-π*跃迁。HL3的最大吸收峰位于412 nm,是HOMO-1到LUMO的跃迁,可归属于邻菲啰啉和三苯胺官能团到硝基官能团的跃迁。338 nm处的吸收峰对应于HOMO到LUMO+3跃迁,可认为是ICT跃迁。279 nm处吸收峰对应于HOMO到LUMO+6,可归属为三苯胺的π-π*跃迁。
| 图表编号 | XD0024456300 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2018.04.20 |
| 作者 | 韦丹蕾、钟卓浩、田玉鹏 |
| 绘制单位 | 安徽大学化学化工学院、安徽大学化学化工学院、安徽大学化学化工学院 |
| 更多格式 | 高清、无水印(增值服务) |

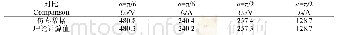



{kind=link}