《表2 分子动力学模拟条件》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《基于密度泛函理论的KF-NaF-AlF_3低温电解质体系势参数拟合及分子动力学模拟》
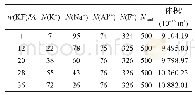
注:w为质量分数;N为粒子数;N(K+),N(Na+),N(Al3+)和N(F-)分别为K+,Na+,Al3+和F-的粒子数;Ntotal为总粒子数。
本文研究的KF-Na F-Al F3熔盐体系化学组成和模拟盒子的体积如表2所示,初始结构模型由Packmol软件[25]随机地将粒子投放到指定大小的盒子中得到。在经典分子动力学模拟中,使用Verlet蛙跳算法求解牛顿运动方程,时间步长设置为1 fs。EWALD求和算法[18]作为处理长程相互作用应用最普遍的方法,在这里被用于处理体系中粒子间的库仑相互作用和偶极相互作用,同时,Buffer宽度设置为0.5×10-10m,能量计算精度为4.184×10-5k J/mol[19]。短程相互作用的截断半径为15×10-10m,离子的电荷设置为+0.67 e(Na),+2.01 e(Al),-0.67 e(F)和+0.67 e(K),这来源于1.2.2节的DFT计算结果。周期性边界条件被用于模拟盒子,用于描述1个没有边界的无限液态系统,使得计算结果更加可靠。模拟盒子系统首先在NPT[24]系综下以0.1 MPa压力升温到827℃,模拟时长为2 000 ps,这意味着模拟过程中保持系统的粒子数(N)、压力(P)和温度(T)为常数。之后,使用NVT系综继续进行3 000 ps结构驰豫。最后,使用Matlab软件统计分析结构驰豫3 000 ps的模拟盒子中的粒子轨迹数据,计算KF-Na F-Al F3熔盐的离子结构。所有的分子动力学计算均在Forcite模块中完成[26]。
| 图表编号 | XD00217070300 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.12.26 |
| 作者 | 王景坤、国辉、张红亮、李劼 |
| 绘制单位 | 中南大学冶金与环境学院、中国宏桥集团有限公司、中南大学冶金与环境学院、中南大学冶金与环境学院、中南大学难冶有色金属资源高效利用国家工程实验室、中南大学冶金与环境学院、中南大学难冶有色金属资源高效利用国家工程实验室 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}