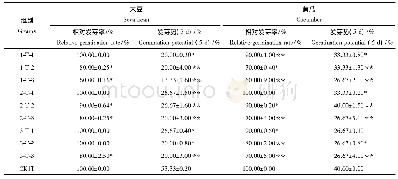
《表1 三联苯系列和甲基联苯系列热力学性质》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《有机质中三联苯成熟度参数及其化学机理:基于地球化学数据和量子化学计算》
本研究的所有密度泛函理论(DFT)计算都采用Becke的三参数交换泛函[30–32],即Lee、Yang和Parr提出的梯度矫正函数结合的密度泛函方法B3LYP。另外,还采用了包含弥散和极化函数的基组6-311++G(d,p),此基组在类似的研究体系中得到了成功应用[28,33–35]。在气相条件下,采用B3LYP/6-311++G(d,p)方法对三联苯系列和甲基联苯系化合物进行了结构优化和频率计算,并确保每一个优化结构没有虚频,得到的结构即为势能面上的极小值。在298.15 K和101325 Pa条件下获得了三联苯系列和甲基联苯系列各异构体的热力学量,如键长、键角、零点振动能(Zero-point Vibrational Energy,ZPVE)、电子能(E)、内能(U)、焓(H)和吉布斯自由能(G)等参数。为直观反映同分异构体之间的热稳定大小,本文中所有热力学能都是以能量最低构型的能量为基准的相对值:将能量最低构型的相对能量定义为0 J/mol,其他分子的绝对能量减去能量最低构型的绝对能量得到相应的相对能量值,如表1所示。相对能量值越大,表明此结构在同分异构体中越不稳定。此外,通过频率计算和分析可以得到任意温度下各个异构体的热力学量。本文所有分子结构都是通过GaussView 5.0软件包实现,并采用Gaussian 09软件包对每一个分子结构进行DFT计算。
| 图表编号 | XD00150257100 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.03.26 |
| 作者 | 刘晓强、李美俊、唐友军、朱志立、祁灵、师生宝、冷筠滢、韩秋雅、肖洪 |
| 绘制单位 | 中国石油大学(北京)油气资源与探测国家重点实验室地球科学学院、四川轻化工大学化学与环境工程学院、中国石油大学(北京)油气资源与探测国家重点实验室地球科学学院、长江大学油气资源与勘探技术教育部重点实验室资源与环境学院、长江大学油气资源与勘探技术教育部重点实验室资源与环境学院、中国石油大学(北京)油气资源与探测国家重点实验室地球科学学院、中国石油勘探开发研究院、中国石油大学(北京)油气资源与探测国家重点实验室地球科学学院、中国石油大学(北京)油气资源与探测国家重点实验室地球科学学院、中国石油大学(北京)油气资 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}