《表2 GGA和GGA+U条件下锶铁氧体的Fe原子磁矩及晶胞总磁矩(μB)》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《Mn掺杂锶铁氧体SrFe_(12)O_(19)电子结构及磁性的第一性原理研究》
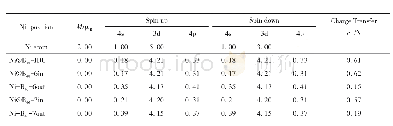
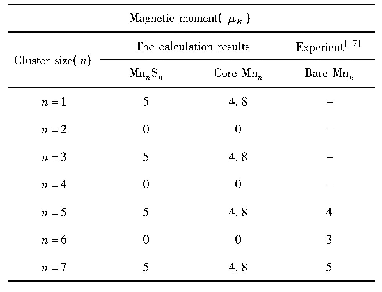
值得注意的是,前述采用GGA方法比较了不同铁磁和亚铁磁结构分布,获得了锶铁氧体稳定磁结构。但由于Fe原子含有高简并度的d电子轨道,电子间的自相互作用较强,需要考虑U值进行修正。基于之前文献的报道结果[28],我们对Fe原子的d轨道考虑U=3.7 eV的修正。研究结果表明,稳定的磁结构与GGA的计算结果相吻合,但是结构和电子性能存在一定差异。对于稳定的磁结构,其晶格参数较不加U时有显著增加,其水平面内以及垂直方向的晶格参数分别由0.586和2.310 nm增加到0.594和2.319 nm。含U值计算得到的晶胞参数与实验测量值a=0.588 nm,c=2.304 nm更吻合[32],表明加U计算对于锶铁氧体体系是必要的。此外,表格2中给出了基于两种方法得到具体的原子磁矩。不加U时,不同位置Fe原子磁矩在3.17~3.73μB。加U后,单个原子磁矩有较大幅度的增加,磁矩范围为4.05~4.18μB,其中12k和2a位置磁矩略高于其他位置,晶胞的总磁矩为40μB。对于电子结构的研究,图2分别给出了锶铁氧体不加U和加U条件下的电子态密度和能带结构。不加U情况下,能带结构和态密度如图2(a~c)所示,可以看出锶铁氧体呈现金属性,与实验值测得的高电阻率明显不符[33]。加U后,锶铁氧体呈现半导体特性。从态密度图2(d)中可以看出,对于占据态来说,12k、2a、2b具有较大的自旋向上电子态,而4f1和4f2具有较大的自旋向下电子态,这与锶铁氧体亚铁磁构型相吻合。12k和2b处Fe原子以及O原子态密度更接近体系的价带顶,而导带底不同占据位的Fe原子以及O原子的态密度比较接近。整个能量区间内,Fe和O原子的态密度有较大的重叠区间,表明Fe和O原子之间有较强的轨道杂化和成键。通过加U计算,获得的能带图如图2(b),自旋向上带隙值为1.71 eV,自旋向下带隙值为1.82 eV,与实验测得的高电阻率较为符合。通过比较加U和不加U条件下晶格参数和电子结构的计算,发现加U计算与实验结果更接近。所以后续掺杂体系的研究都采用GGA+U的方法。
| 图表编号 | XD00112183400 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.10.01 |
| 作者 | 王中、查显弧、吴泽、黄庆、都时禹 |
| 绘制单位 | 哈尔滨理工大学材料科学与工程学院、中国科学院宁波材料技术与工程研究所先进能源材料工程实验室、中国科学院宁波材料技术与工程研究所先进能源材料工程实验室、哈尔滨理工大学材料科学与工程学院、中国科学院宁波材料技术与工程研究所先进能源材料工程实验室、中国科学院宁波材料技术与工程研究所先进能源材料工程实验室 |
| 更多格式 | 高清、无水印(增值服务) |

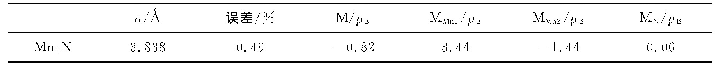

{kind=link}