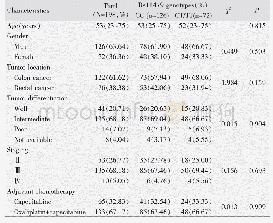
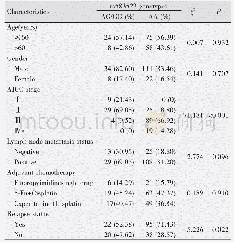
《Table 4 Allele discrepancy of rs506121,rs2071676,rs11040923 between cases and controls.》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《New Gene Variants Associated with the Risk of Chronic HBV Infection》
P values less than 0.05 in meta-analysis are indicated in bold DOCK8 Dedicator of cytokinesis 8;CA9 carbonic anhydrase 9;DNHD1 dynein heavy chain domain 1;ORodds ratio;CI confidence interval.*If the OR was calculated as C/T,then the frequency listed in th
In the first stage,we did not find any potentially functional gene variants that may have a relationship with HBVinfection at the Bonferroni genome-wide significance level(Fig.1).Reasons accounting for this may be as follows:first,exome sequencing only focuses on genetic variation in the exome region,and is thus very likely to neglect those lying in the noncoding region(Wang et al.2018);second,statistical power of classical single-variant based association tests for low-frequency and rare variants is low when the sample sizes are small(Lee et al.2014;Wang et al.2018).In the following stage,we expanded our sample sizes and further conducted a TOF analysis to confirm the potential gene variants found in the discovery-phase.Twostage design is the standard strategy of genome-wide association study for discovering and validating the candidate genetic makers.Stage 2 is a replication study which focuses on the statistical significance discovered in stage 1.Previous study demonstrated that joint analysis of replication-based study result in increased power to detect genetic association(Skol et al.2006).And for evaluating the significance of association between genetic factors and affected-unaffected phenotypes,Chi square test,Fisher’s exact test(Purcell et al.2007)and mixed regression model(Zhou and Stephens 2012)are the most popular methods designed for population matched and un-matched studyrespectively.In our study,we used Chi square test to perform the joint analysis on 30 variants in two populations.Combining the results of these two stages,we observed that cases in CHB group have significantly higher frequency of DOCK8-T allele and higher frequency of CA9—A allele than controls in clearance group(Table 4).
| 图表编号 | XD00159092200 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.08.01 |
| 作者 | Mengjie Fan、Jing Wang、Sa Wang、Tengyan Li、Hong Pan、Hankui Liu、Huifang Xu、Daria V. Zhernakova、Stephen J. O’Brien、Zhenru Feng、Le Chang、Erhei Dai、Jianhua Lu、Hongli Xi、Yanyan Yu、Jianguo Zhang、Binbin Wang、Zheng Zeng |
| 绘制单位 | Department of Infectious Diseases, Peking University First Hospital、Department of Medical Genetics and Development Biology,School of Medical Basic, Capital Medical University、Center for Genetics, National Research Institute for Family Planning、Department |
| 更多格式 | 高清、无水印(增值服务) |
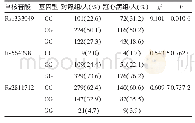
![表4 两组UCP2基因rs757747279、rs45541732和rs660339位点基因型分布[例(%)]](http://bookimg.mtoou.info/tubiao/gif/SYLC202019009_03000.gif)

![Table 5 An rs4944831-rs1783596-rs4944832 haplotype analysis[n (%) ]of the P2RY2 gene in ischemic stroke patients and con](http://bookimg.mtoou.info/tubiao/gif/SJZY201903021_15300.gif)
{kind=link}