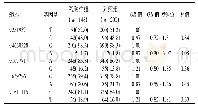
《表3 肺念珠菌感染组与对照组在4种遗传模型下基因型分布》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《贵州汉族慢性阻塞性肺疾病患者Toll样受体1基因多态性与肺念珠菌感染及定植的相关性》
为避免人群分层影响,将性别与年龄作为协变量进行校正后计算OR值与95%CI。TLR1基因rs4833095在共显性、显性及超显性遗传模式下在肺念珠菌感组与对照组间基因型分布的差异均有统计学意义(P<0.05)。与对照组相比,在共显性遗传模式下,以野生型CC为参照,CT基因型是肺念珠菌感染的风险基因型(OR=2.48,95%CI:1.36~4.54,P=0.0099);显性遗传模型下,CT+TT突变型携带者较CC野生纯合型携带者肺念珠菌感染的发病风险增高(OR=2.36,95%CI:1.33~4.20,P=0.0028);在超显性遗传模式下,以纯合基因型CC+TT为参照,杂合性携带者CT是肺念珠菌感染的危险因素(OR=2.08,95%CI:1.20~3.60,P=0.0084)。根据最小AIC与BIC值,推测该位点最佳遗传模式是显性;而TLR1基因rs5743618在肺念珠菌感染组与对照组间基因型频率的差异无统计学意义(P>0.05),见表3。两位点在念珠菌定植组与对照组间基因型频率差异均无统计学意义(P>0.05),见表4。此外,采用SNPstats在线软件对rs4833095与rs5743618进行连锁不平衡分析,发现这两个SNP之间不存在连锁不平衡,故不进一步做单倍型分析。
| 图表编号 | XD0035254300 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.04.01 |
| 作者 | 刘海文、彭春红、林洁如、张湘燕 |
| 绘制单位 | 贵州医科大学研究生学院、贵州省人民医院呼吸与危重症医学科、贵州省人民医院呼吸与危重症医学科、贵州省人民医院呼吸与危重症医学科 |
| 更多格式 | 高清、无水印(增值服务) |




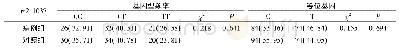
{kind=link}