《表2 单一疾病预测模型的测试结果》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
在2016年,Pasolli等[25]提出了Met AML(Metagenomic Rediction Analysis Based on Machine Learning)预测工具,利用Met AML构建单一疾病预测模型的基本步骤包括(图3A):(1)对宏基因组数据按照人类微生物组项目的标准操作规程进行预处理,并删除某些仅含少量核苷酸的Reads (如:对于IBD数据集最小核苷酸个数为70)。(2)采用默认参数的Meta Phl An2[53]方法进行特征提取,获得物种水平的相对丰度和特异菌株标记两种特征。(3)分别基于SVM、RF等算法建立预测模型。(4)利用10倍交叉验证对所构建模型的性能进行评估测试,结果见表2。该模型只能预测一种疾病发病情况(由训练数据集决定),本质上是一个二分类模型,因此模型的适用性较差,而其优势在于预测精度高、模型构建过程简单。
| 图表编号 | XD00210330000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2021.01.20 |
| 作者 | 李强、衣杨、吴忠道、丁涛 |
| 绘制单位 | 中山大学数据科学与计算机学院、中山大学数据科学与计算机学院、中山大学新华学院信息科学学院、中山大学中山医学院、中山大学中山医学院 |
| 更多格式 | 高清、无水印(增值服务) |

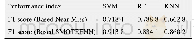




{kind=link}