《表1 FBTAK和FPTAK的分子轨道能量和极化率》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
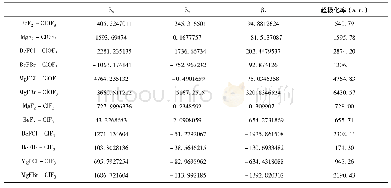
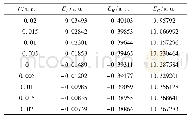
为探索苯并噻吩和苯联噻吩两种结构单元对分子中电子云分布、电子云被极化的难易程度的影响并进一步探索与三阶NLO性质的关系,运用Gaussian 09量子化学程序包[22],采用密度泛函理论方法在B3LYP/6-311+G(d,p)水平上优化计算两化合物分子的几何结构、极化率、HOMO、LUMO结构(图3)。通过计算分析获得FBTAK和FPTAK的分子轨道能量和共轭电子体系的极化率(表1)。
| 图表编号 | XD00106319800 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.11.18 |
| 作者 | 王桂林、孙金鱼、尹爱萍、王迎进、赵明根 |
| 绘制单位 | 忻州师范学院化学系山西省材料与计算化学重点实验室、忻州师范学院化学系山西省材料与计算化学重点实验室、忻州师范学院化学系山西省材料与计算化学重点实验室、忻州师范学院化学系山西省材料与计算化学重点实验室、忻州师范学院化学系山西省材料与计算化学重点实验室 |
| 更多格式 | 高清、无水印(增值服务) |
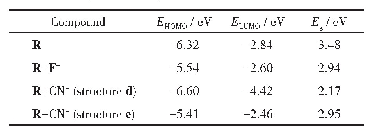


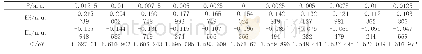
{kind=link}