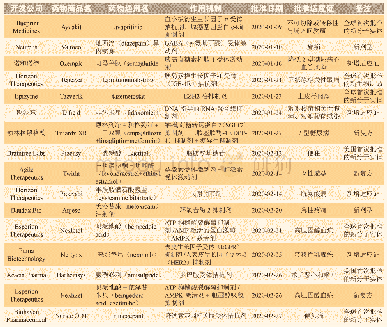
《表2 美国FDA再生医学治疗相关指导原则》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
美国FDA采用经典的“法规-监管-指导原则”三级监管体系.其下属的生物制品评价与研究中心(center for biologics evaluation and research,CBER)专门设立了组织和先进疗法办公室(office of tissues and advanced therapies,OTAT)对组织、细胞和基因治疗产品进行监管,形成了20余年的再生医学产品监管经验[14].法规层面,美国公共健康服务法案(PHS Act361)第361部分针对自体来源、最小化处理和同源使用的组织细胞产品,第351部分针对异体来源、复杂处理和非同源使用的治疗性细胞产品[15].2005年,美国21世纪联邦法案中1271部分(21 CFR 1271)生效,详细规定了组织细胞产品注册、供者筛查、生产过程控制、组织操作规范等内容.人体细胞、组织和细胞/组织治疗产品在21 CFR 1271中定义为含有或由用于植入、移植、输注或转移到人类受体的人体细胞或组织组成的物质.按照风险等级,HCT/Ps被分为需要和不需要上市审批两种类型:仅受361规定的HCT/Ps产品不需要上市前审批,但需上市前在FDA注册,并证明该产品不会导致传染病传播,在临床试验开展过程中FDA也将密切关注,随时对存在非最小化处理和非同源使用的临床试验进行监督[16].符合PHS Act 351规定范围的产品必须按照研究型新药申报临床试验(IND),并需上市前审批(BLA)[17].考虑到细胞产品的安全性,20多年来CBER针对供体筛选、移植手术操作、不同组织类型(血液、胰岛、心脏、软骨等)、适应症、偏差处理等各方面都制定了详细的指导原则综合来看,CBER着重考察干细胞产品的供体来源、原材料质量、生产工艺过程控制和产品的深度质量分析.FDA近期针对再生医学治疗发布的指导原则和概要见表2.
| 图表编号 | XD0034728600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.01.20 |
| 作者 | 卢加琪、刘伯宁、罗建辉 |
| 绘制单位 | 国家药品监督管理局药品审评中心、国家药品监督管理局药品审评中心、国家药品监督管理局药品审评中心 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}