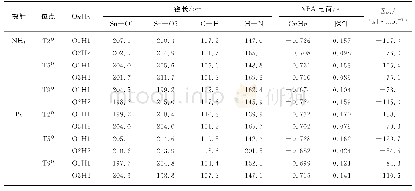
《表1 NH3在Ir1/MoS2体系上吸附不同构型的几何参数》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《单原子催化剂Ir_1/MoS_2表面上NH_3吸附理论研究》
图2-A中NH3分子通过N原子和Mo S2表面上的Ir原子相连,它的C3垂直于体系Ir1/Mo S2,三个N-H键指向远离表面的方向,且投影至Ir-S键方向.图2-C也为垂直结构,但三个N-H键是投影至六角表面的fcc位,图2-A和图2-C两种构型吸附能均为1.58 e V,计算结果如表1所示.Ir-N和N-H键长(dIr-N、dN-H)以及夹角∠HNH也几乎相同,与气相NH3构型(dN-H=0.102 nm,∠HNH=107.3°)比较无明显变化,可见吸附之后NH3分子结构没有发生明显改变,说明内层轨道在吸附过程中受的扰动非常小,基本保持了其在气态分子中的特征.
| 图表编号 | XD00200103900 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2021.02.28 |
| 作者 | 肖香珍、银召利、张建伟 |
| 绘制单位 | 河南科技学院实验管理中心、河南科技学院新科学院、河南科技学院实验管理中心 |
| 更多格式 | 高清、无水印(增值服务) |



![表1[9]合成氨反应在不同压强下的平衡体系中NH3的含量](http://bookimg.mtoou.info/tubiao/gif/HZSB202005026_08200.gif)

{kind=link}