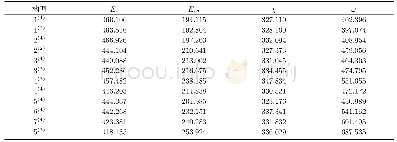
《表1 团簇Co3Ni B2 12种优化构型的能隙差》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
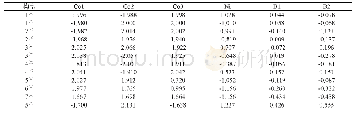
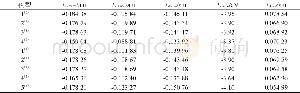
20世纪50年代,日本化学家福井谦一提出前线轨道理论,依据是在团簇分子中,轨道电子能量最高、所受束缚力最小、电子最活泼且最容易发生变动的HOMO轨道与在所有未占轨道中能量最低、最容易接受电子的LUMO轨道一同决定着分子的电子得失和转移能力,同时还决定着分子间反应的空间取向等重要化学性质.表1列出了团簇Co3Ni B2各优化构型的HOMO、LUMO轨道能量以及能隙差的具体数值.HOMO轨道能量EHOMO越高,说明团簇分子给出电子的能力越强;LUMO轨道能量ELUMO越低,说明其接受电子的能力越强.为了更清晰地了解电子从占据轨道向空轨道跃迁的能力,引入能隙差EGAP(EGAP=ELUMO-EHOMO)来表征团簇在与小分子参加化学反应时的化学反应活性.能隙差值越小,电子越容易跃迁,团簇分子就越容易参与化学反应,反应活性就越强;反之,能隙差值越大,电子跃迁越不容易,团簇分子就越不易参与化学反应,反应活性就越弱.
| 图表编号 | XD00198449400 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.11.01 |
| 作者 | 秦渝、方志刚、张伟、廖薇、许友 |
| 绘制单位 | 辽宁科技大学化学工程学院、辽宁科技大学化学工程学院、辽宁科技大学化学工程学院、辽宁科技大学化学工程学院、辽宁科技大学化学工程学院 |
| 更多格式 | 高清、无水印(增值服务) |



{kind=link}