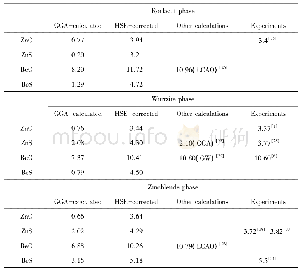
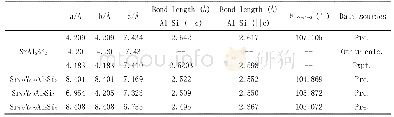
《表1 GGA和GGA+U两种方法结构优化后Zn1-xMgxO (0≤x≤0.25) 合金晶胞参数Tab.1 Geometry optimization results of different do
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《Zn_(1-x)Mg_xO电子结构及光学性质的第一性原理GGA+U方法研究》
本文计算采用的是基于密度泛函理论(DFT)框架下的CASTEP软件包[15]。电子间交换关联能用广义梯度近似的PBE[16]泛函描述,电子和离子间的相互作用采用超软赝势,几何优化法采用了BFGS算法[17]。系统总能量和电荷密度在布里渊区的计算使用Monkhorst-Pack[18]方案来选择型k空间格点,布里渊区k点的选取分别为4×4×2,4×4×2,4×4×4,4×4×5,对应于x为0,0.062 5,0.125,0.25的超晶胞模型。计算在不固定任何参数下进行几何优化,能量、自洽场和能带的收敛精度皆设为1.0×10-5e V/atom,平面波截断能为480 e V,原子间的相互作用收敛精度为0.5 e V/nm。采用GGA和GGA+U两种方法优化得到晶格参数,见表1,优化后的晶格常数均比实验值略大,但误差都小于1.5%,且后者计算所得的结果更接近实验值。由表1可知,ZMO晶格常数a值随x增大而增大,c值随x增大而减小,变化趋势与实验结果[5]相一致。
| 图表编号 | XD0017630700 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2018.06.01 |
| 作者 | 何旭、武莉莉、任胜强、张静全、都政 |
| 绘制单位 | 四川大学材料科学与工程学院、成都纺织高等专科学校、四川大学材料科学与工程学院、四川大学材料科学与工程学院、四川大学材料科学与工程学院、国家超级计算深圳中心 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}