《表4 配合物的L-J体系极小能随α(1)和α(2)旋转角变化》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《"三(2,4-二氯苄基)氯化锡二甲基甲酰胺配位聚合物的合成和结构"》
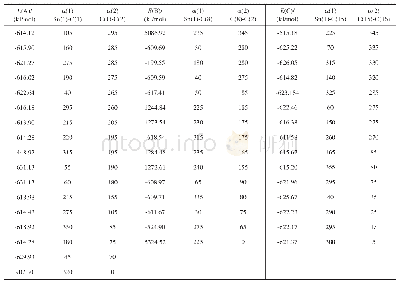
以晶体分子结构为初始构象,设所考察的单键发生旋转时,其它原子或基团不发生位移,考察锡-碳(氧)键及该碳与苯环单键旋转(为了简化计算,取每旋转5°,计算一个单点能)形成空间各个构象的L-J作用能与旋转角的关系[11]。设Sn(1)-C(1)键旋转角为α(1),C(1)-C(2)键旋转角为α(2),体系能为E(A);Sn(1)-C(8)键旋转角为α(1),C(8)-C(9)键旋转角为α(2),体系能为E(B);Sn(1)-C(15)键旋转角为α(1),C(15)-C16)键旋转角为α(2),体系能为E(C),产生三维体系能随旋转角[α(1),α(2)]变化。配位DMF分子向Cl-Sn键的反向进入与锡配位产生O(1)→Sn(1)后,由O(1)-Sn(1)键旋转α,DMF将产生空间影响,体系的二维能量E(D)随α[O(1)-Sn(1)]的变化曲线,如图4(D)所示。DMF的作用在相对初始结构160°和280°时分别存在两个极大能量值,最大能垒为208.30 k J/mol,可见,DMF空间影响不大,另外,DMF的空间效应还有三个能量极小点:在相对初始结构68°、220°和349°时,E(D)分别为-613.98,-595.02和-603.34 k J/mol,因此,DMF在晶体结构中影响较小。从图4的E(A)~E(C)看出,体系能随α(1)和α(2)变化的能值为曲面,其中存在多个极大能,表明苄基的空间作用有着明显的影响,且三个2,4-二氯苄基的影响也不同,它们的最大能峰值分别为E(Amax):8 899.94 k J/mol、E(Bmax):22 822.49 k J/mol和E(Cmax):1 185.24 k J/mol,可见,处于Z式的2,4-二氯苄基产生极高的能垒,为极不稳定的一种结构。处于同向的两个2,4-二氯苄基之间的这一作用能相对较低,第三个苄基[环C(16)~C(21)]产生的能垒较小,这从理论上支持了三(苄基)氯化锡的螺旋桨结构[4]。从图4和表4还可见出,配合物的三个2,4-二氯苄基的空间构型还存在17个E(Amin)和15个E(Bmin)、E(Cmin)极小能,这些低能构象中,相对结构的初始旋转角,最低体系能在α(1)和α(2)分别为55°、190°和60°,180°时,E(Amin):-631.13 k J/mol;分别为50°、185°时,E(Bmin):-617.41 k J/mol;分别为315°、330°时,E(Cmin):-626.05 k J/mol。进一步证明,处于Z式2,4-二氯苄基的低能构象较高于另两个苄基引起的空间影响大,最高体系能与最低能差ΔE(B)>ΔE(A)>ΔE(C),均得一致的空间影响,为有机锡配合物的配位研究提供有益的参考。
| 图表编号 | XD00149205900 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.06.15 |
| 作者 | 邝代治、张复兴、杨春林、朱小明、谭宇星 |
| 绘制单位 | 衡阳师范学院化学与材料科学学院功能金属有机化合物湖南省重点实验室功能金属有机材料湖南省普通高等学校重点实验室、衡阳师范学院化学与材料科学学院功能金属有机化合物湖南省重点实验室功能金属有机材料湖南省普通高等学校重点实验室、衡阳师范学院化学与材料科学学院功能金属有机化合物湖南省重点实验室功能金属有机材料湖南省普通高等学校重点实验室、衡阳师范学院化学与材料科学学院功能金属有机化合物湖南省重点实验室功能金属有机材料湖南省普通高等学校重点实验室、衡阳师范学院化学与材料科学学院功能金属有机化合物湖南省重点实验室功能金 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}